Five subjects are being tackled:
1- Physiopathological mechanisms associated with cytosolic or nuclear intermediate filaments and their partner proteins in myopathies:
A/ Myopathies related to nuclear protein (head B.Buendia): Nathalie VADROT and Dr Brigitte BUENDIA.
Mutations in the LMNA gene encoding A-type lamins, nuclear proteins belonging to the intermediate filament proteins, are responsible for diverse diseases. We are studying their underlying molecular mechanisms. Our collaboration with clinicians (team of Pr C.Vigouroux) and development of animal and cellular models, allowed us to reveal that A-type lamins mutated at some sites responsible for myopathies had a different capacity than wild-type proteins to assemble, bind DNA and/or protein partners, with consequences on nuclear morphology (Figure 1) and the 3D organisation / expression of the genome, associated with defects of cell proliferation /differentiation or muscle fiber type. Our present project focuses on one partner of A-type lamins, LAP2, from which our colleagues (team of Dr P. Richard) just identified few mutants associated with pathologies of the cardiac muscle.
Publications of the year: Paulsen et al. 2017; Barateau et al. 2017
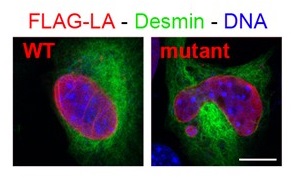
Figure 1: Immunofluorescence of myoblasts expressing FLAG-tagged lamin A (LA) either wild-type or mutant (responsible for congenital muscular dystrophy); from Barateau et al. 2017.
B/ Desmin-related myopathies (desminopathies) (head: S.Batonnet-Pichon) : Pr Patrick VICART, Dr Sabrina PICHON, Dr Alain LILIENBAUM, Dr Eva CABET, Florence DELORT, Coralie HAKIBILEN.
Desminopathies are rare diseases affecting the neuromuscular system. They are usually transmitted in an autosomal dominant mode and appear most often in adulthood. They are particularly characterized by the aggregation of desmin, an intermediate filament located in the sarcoplasm of skeletal and cardiac muscle cells. To date more than seventy mutations associated with this pathology have been described on the desmin gene. Our laboratory has historically participated in the identification of at least twenty of them. Currently, we are studying the more specifically missense mutations D399Y, E413K, R406W (Figure 2). Our approaches are highly multidisciplinary, which allows us to enrich our prospects of desmin studies.
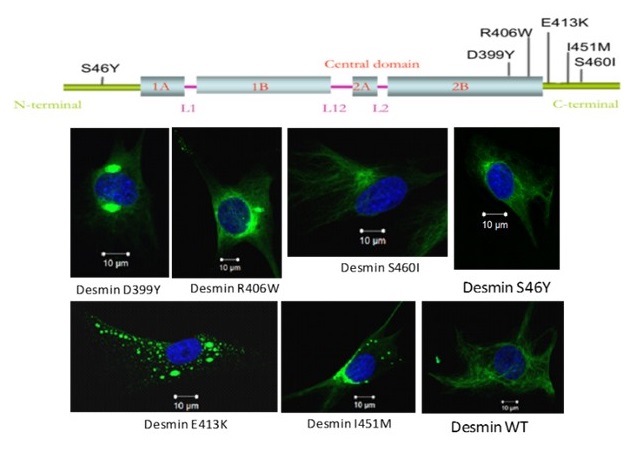
Figure 2: Schematic representation of desmin protein and the localization of the main mutations studied in the laboratory
Thus, in order to study the molecular mechanisms linked to desminopathies, we have developed various models ranging from stable clones derived from C2C12 myoblasts, expressing wild or mutated inducible desmin, to animal models with AAV injection (# 5441 CEF Ethics Committee) or more recently a R405W Knock-in mouse (murine homologous to R406W human mutation, CEB-16-2016 Ethics Committee). Using these models, we therefore explore different molecular mechanisms related to these pathologies such as:
a- First, we have chosen to study a mouse model of myofibrillar myopathy in order to better understand the early stages of the disease development and to evaluate therapeutic strategies. The R406W human desmin mutation produces severe and early effects in patients, especially on, cardiac, respiratory and the limbs skeletal muscles. The knock-in mouse allows the endogenous gene to be replaced by the mutated gene and is the most physiological approach. The hetero- and homozygous mice show an abnormal accumulation of desmin at the sarcoplasmic membrane of the striated muscles, in particular in the Tibialis Anterior and Soleus muscles. This accumulation worsens with age. In addition, homozygous mice are smaller and die at about 3 months of age. Studies are under way to clarify whether this lethality is due to cardiac, respiratory or other critical defects in other organs (Figure 3).
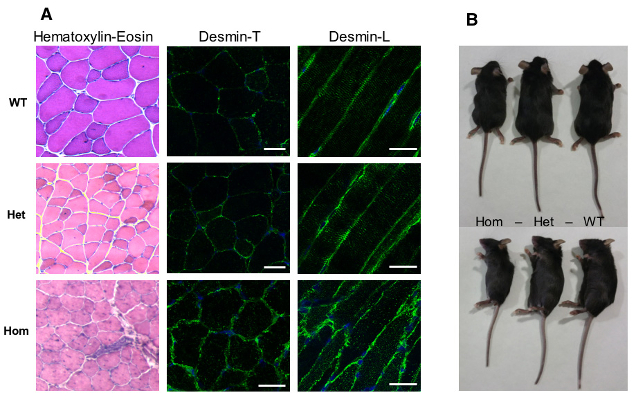
Figure 3: Model of murine desR405W Knock-in (homologous to R406W human mutation): homozygous mice present a structural alteration of muscular fibres and a significant size reduction.
b- Second, we are studying the mechanical properties of the cells expressing wild or mutated desmin thanks to two strong collaborations established with biophysicists within an ANR partnership in progress:
- F. Briki (LPS, Orsay): we have shown that the expression of the D399Y desmin mutant shows an alteration of the reorientation under stress (Leccia et al., 2013). We have also studied the rigidity of cells expressing desmin D399Y using atomic force microscopy (Article under review)
- S.Henon (MSC, Paris Diderot): we have recently shown that overexpression of wild-type desmin by electroporation in C2C12 is able to globally rigidify the cells. On the contrary, the E413K mutation does not induce this stiffening assuming loss of function. In addition, using the Single Cell Rheometer (A. Asnacios, MSC, Paris Diderot) and Traction Force Microscopy (B.Kalman, Grenoble), we have determined that the overexpression of desmin E413K in C2C12 decreases their ability to generate contractile force (Charrier et al, 2016). This effect is associated with an alteration in the number and length of actin stress fibers (Charrier et al., 2016). Finally, we have also shown that desmin plays a role in the cytoplasmic rheological properties of cells and not cortical ones, using optical and magnetic tweezers (Charrier et al., Submitted).
c- More recently, with the arrival of C.Hakibilen (PhD student of BioSPC), still in collaboration with S.Hénon (MSC Paris Diderot), we have started a new project (AFM funding) on the desmin role and the impact of its mutations on the cell-matrix adhesion (Figure 4, left), and on the cell-cell adhesion (collaboration being set up with RM Mège, IJM, Paris Diderot). This project is based in part on the use of Total Internal Reflection Fluorescence Microscopy (BFA) imaging, allowing us to more specifically quantify focal adhesions (their number, area …). These analyzes will be carried out not only on C2C12 (Figure 4, right), but also on primary satellite cells derived from the desKO and R405-KI mice avaible in the laboratory
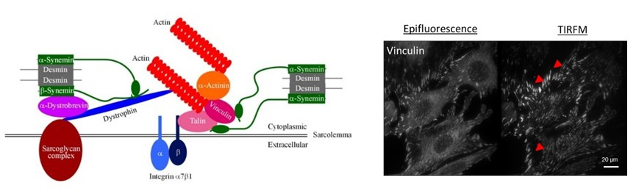
Figure 4: At left, schematic representation of desmin protein in a focal adhesion in cardiac cells. At left, vizualisation of focal adhesions of C2C12 cells coated on fibronectin with epifluorescence or TIRFM (BFA, plateau technique) imaging.
d- From a biochemical point of view, we are also studying the interactions with different partners such as the Dux 4 and Dux4C proteins in collaboration with F. Coppée (Mons, Belgium).
e- We have also demonstrated that oxidative or thermal stress induces the appearance of D399Y desmin aggregation in cellulo (Segard et al., 2013, Figure 5). This effect has been confirmed for other mutants such as Q389P, or R406W but cannot be extended to all mutations of desmin or of the 2B region (Delort F. et al., Article in redaction).
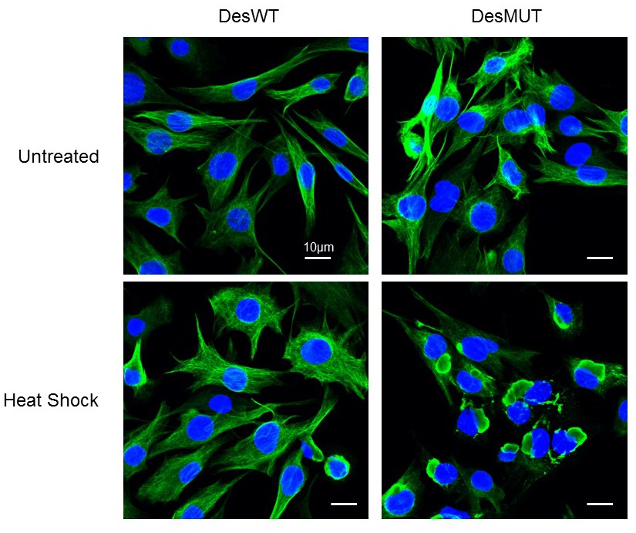
Figure 5: Effect of a Heat-shock treatment on D399Y desmin aggrégation in inducible stable clones. (Image taken with the LSM700 confocal microscope of the ‘plateau d’Imagerie BFA’)
f- Finally, knowing that at present no treatment is available for patients with desminopathies, we are actively looking for molecules with therapeutic potential. Thus the knowledge gained through our work and our different models allowed us to test different molecules such as antioxidants such as N-acetyl-L-cysteine (NAC) (Segard et al., 2013) or tocopherols (Cabet E and Al, 2015) but also pro-autophagic molecules such as pp242 (Cabet E. et al, 2015) (Figure 6). We have thus been able to demonstrate in cellulo that a pretreatment with these molecules decreases the appearance of the aggregation of desmin mutants. These results are currently being studied on the animal model (AAV and DesKI) but open interesting therapeutic perspectives.
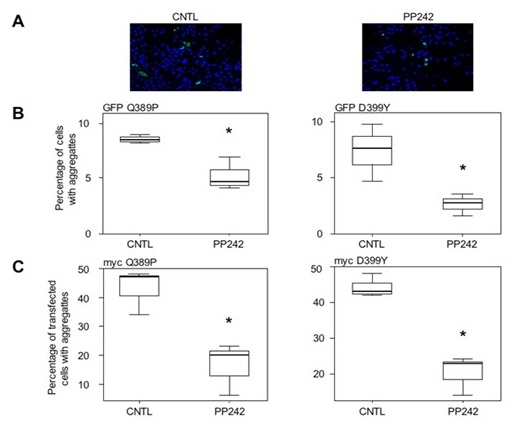
Figure 6: Effect of pp242 treatment on D399Y and Q389P desmin aggregation (Image taken with the inverted ZEISS microscope of the ‘plateau d’Imagerie BFA’)
2- Congenital Myopathies (Head: A.Ferreiro):
A/ SEPN1-related myopathies: Dr Ana FERREIRO, Dr Anne FILIPPE, Dr Maryline MOULIN, Nathalie VADROT, Swati DUDHAL.
We have an internationally acknowledge expertise in the field of early- onset myopathies (EOM), which are heterogeneous, inherited muscle disorders presenting with muscle weakness from birth or early infancy. We develop a multidisciplinary approach to the study of EOM, situated at the interface clinical/basic research.
In the last years, thanks to a collaborative strategy, we defined a line of research on EOM, created and animated a European network which provided a critical mass of patients, identified that mutations in 4 genes are responsible for 8 forms of EOM, reassessed the corresponding phenotypes and contributed to establish a new, genetically-based nosological classification in this field.
Currently, we focus on getting a better knowledge of the mechanisms leading from the genetic defects in EOM to the phenotypical consequences in patients, in order to develop pathophysiology-based therapeutic approaches, namely pharmacological therapies. Particularly, we established or contributed to establish that mutations of the SEPN1 gene, encoding selenoprotein N (SelN), are responsible for 4 categories of EOM.
Using an exvivo model (cultured cells from patients), we have recently established that oxidative/nitrosative stress is implicated in the pathophysiology of SEPN1-related myopathy, and that antioxidants are an effective ex-vivo treatment. Oxidative stress induces epigenetic modifications in stem cells, and preliminary data obtained by other groups suggest a modulation of satellite cell markers by SelN. We are interested in characterizing further these mechanisms, and also the mechanistic links between oxidative stress, the severe muscle atrophy observed in patients with SEPN1 mutations and the modulation of expression of genes involved in cell survival and death.
We will develop additional pharmacological studies ex-vivo and on the animal model of SelN deficiency, to analyze the effects of antioxidants on the pathways mentioned above. A first clinical trial of SEPN1-related myopathy with antioxidants is being designed.
B/ ASC-1 related myopathy (TRIP4): Dr Ana FERREIRO, Dr Isabelle DUBAND-GOULET, Dr Eva CABET, Dr Alain LILIENBAUM.
C/ TTN-related myopathies: Dr Ana FERREIRO.

